You can view the slides from my presentation here.
Required Modules
Data Preprocessing
Taxonomic Classification
Functional Analysis
Visualization
srun --mem 15GB --cpus-per-task 4 --pty /bin/bash
module purge all
module load gencore/1 gencore_metagenomics
Input Data
Since the databases we will be querying are large, we will create symbolic links (pointers) to the data instead of copying them.
cd $SCRATCH
mkdir metagenomics
cd metagenomics
ln -s /scratch/gencore/datasets/metagenomics/demo.fastq
ln -s /scratch/gencore/datasets/metagenomics/SRS014459-Stool.fasta.gz
ls /scratch/gencore/datasets/metagenomics/Homo_sapiens_Bowtie2_v0.1/Homo_sapiens.* | xargs -I {} ln -s {}
ln -s /scratch/gencore/datasets/metagenomics/minikraken_20141208
Introduction
Metagenomics is the study of genetic material recovered directly from environmental samples. This is useful when attempting to understand what microbes are present and what they are doing in a microbiome, such as soil or the human gut. Given that these studies are often aimed at sequencing a population of diverse microbes that vary in abundance, along with many inherent biases, this is arguably one of the most complex types of datasets in current use. A useful tactic to analyzing metagenomic datasets is to simplify the problem: remove the noise, extract data that you know, and attempt to classify what you don't. With this strategy, you are reducing the complexity of the dataset, therefore making your analysis easier and more intuitive. This tutorial will provide a brief introduction on how to asses what you have in your sample and provide a roadmap to where to go from there.
Overview
There are dozens of tools available to help analyze metagenomic datasets. However, there are some common questions and workflows that are shared in almost all studies:
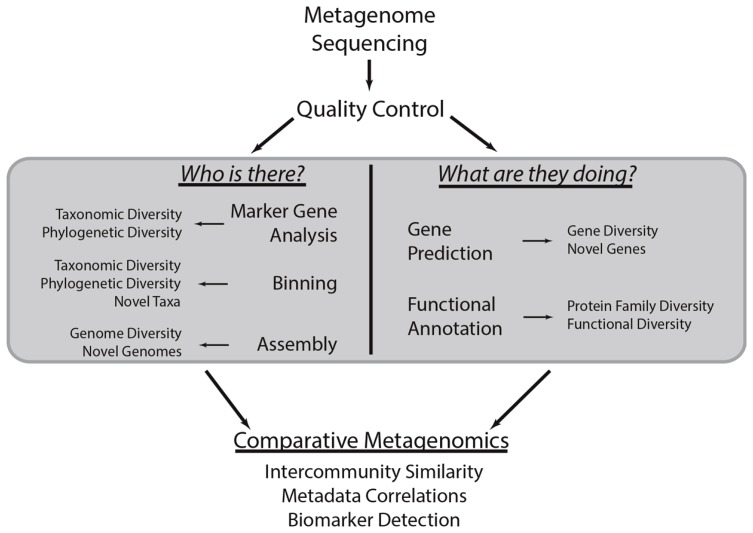
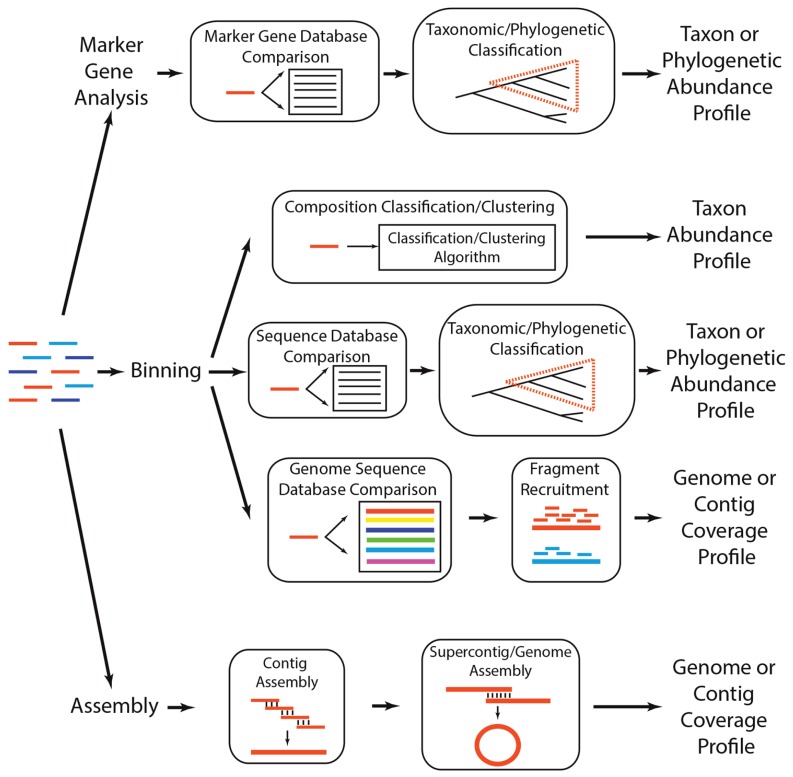
To answer these questions, many different strategies have been employed, but they all start with defining a profile of the organisms within the community. There are several ways to do this, though some of the options are limited to the type of data that you have. There are essentially two types of metagenomic datatypes: targeted and shotgun. Targeted usually focuses in on a select number of loci, for example the 16s rRNA gene in bacteria. Shotgun is unrestricted and aims at sequencing everything in your sample. Two example workflows for each are seen below:
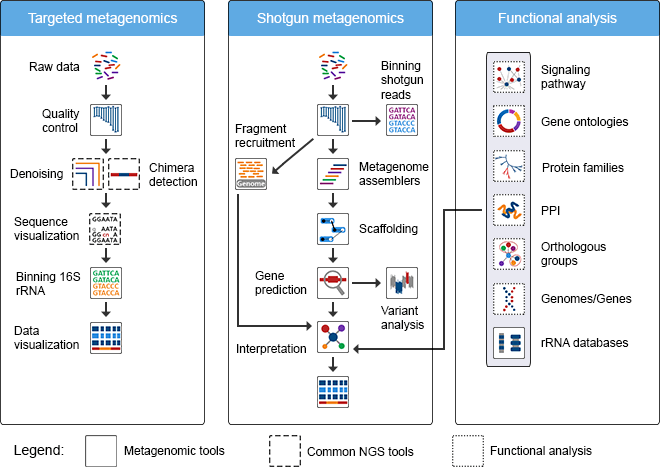
For this exercise we will be looking at the more recent and more relevant shotgun metagenomic analysis, specifically marker gene analysis and binning - assembly is near impossible so we'll save that for another time. There are multiple ways to assess the questions above, and below are the most common practices to identify taxonomy.