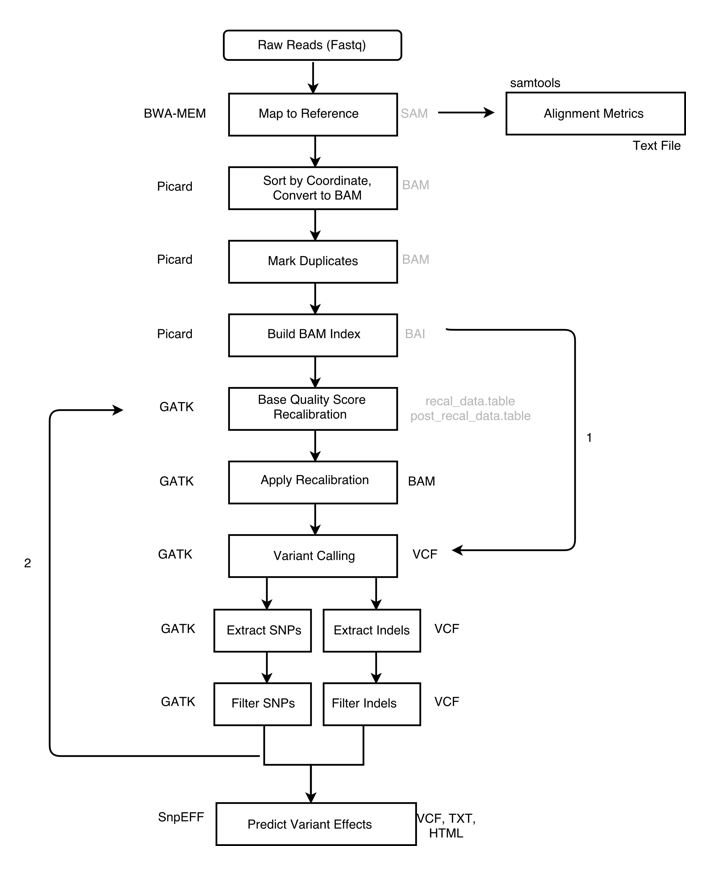
Pre-Processing
Raw data (typically FASTQ files) are not immediately usable for variant discovery analysis. The first phase of the workflow includes the pre-processing steps that are necessary to get your data from raw FASTQ files to an analysis-ready BAM file.
Overview:
- Align reads to reference
- Sort sam file (output from alignment) and convert to bam
- Alignment Metrics
- Mark duplicates
- Prepare reference dictionary, fasta index, and bam index
- Recalibrate base quality scores
Setting up your environment on Dalma
For users working on the Dalma HPC in NYUAD :
1) Enter an interactive Slurm session
srun --mem 10GB --cpus-per-task 4 --pty /bin/bash
2) Load gencore and variant detection modules
module load gencore/1
module load gencore_variant_detection
3) Get sample dataset
cd /scratch/<netID>/
cp /scratch/gencore/datasets/variant_calling.tar.gz .
tar xvzf variant_calling.tar.gz
cd variant_calling
1) Alignment
We will use the BWA MEM algorithm to align input reads to your reference genome. We use BWA MEM because it is recommended in the Broads best practices and because it has been found to produce better results for variant calling. Note that BWA MEM is recommended for longer reads, ie. 75bp and up.
Alternative aligners such as Bowtie2 may be used.
Note: Aligners typically require an indexed reference sequence as input.
If required, index files can be built from a reference sequence (in FASTA format) using the following command:
bwa index <reference.fasta>
Using the reference sequence in the sample dataset, we can build the index files using the following command:
bwa index GCF_000001405.33_GRCh38.p7_chr20_genomic.fna
If executed correctly, you should see the following output:
[bwa_index] Pack FASTA... 0.95 sec
[bwa_index] Construct BWT for the packed sequence...
[BWTIncCreate] textLength=128888334, availableWord=21068624
[BWTIncConstructFromPacked] 10 iterations done. 34753182 characters processed.
[BWTIncConstructFromPacked] 20 iterations done. 64202446 characters processed.
[BWTIncConstructFromPacked] 30 iterations done. 90372990 characters processed.
[BWTIncConstructFromPacked] 40 iterations done. 113629422 characters processed.
[bwt_gen] Finished constructing BWT in 48 iterations.
[bwa_index] 33.57 seconds elapse.
[bwa_index] Update BWT... 0.68 sec
[bwa_index] Pack forward-only FASTA... 0.60 sec
[bwa_index] Construct SA from BWT and Occ... 11.97 sec
[main] Version: 0.7.8-r455
[main] CMD: bwa index GCF_000001405.33_GRCh38.p7_chr20_genomic.fna
[main] Real time: 48.246 sec; CPU: 47.800 sec
Let's take a look at the output using ls -l
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.amb
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.ann
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.bwt
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.pac
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.sa
We can see 5 new files, all having the same basename as the original reference sequence file. These are the index files required by BWA.
Note: If the reference is greater than 2GB, you need to specify a different algorithm when building the BWA index, as follows:
bwa index -a bwtsw <reference.fasta>
Once we have the reference index, we can proceed to the alignment step. We run BWA as follows:
bwa mem -M -R <readgroup_info> <ref> <reads_1.fastq> <reads_2.fastq> > <output.sam>
Command explained:
bwa mem Invoke the bwa mem algorithm
-M This flag tells bwa to consider split reads as secondary, required for GATK variant calling
-R <readgroup_info> Provide the readgroup as a string. The read group information is key for downstream GATK functionality. The GATK will not work without a read group tag.
<ref> The name of your reference sequence. Note that all index files must be present in the same directory and have the same basename as the reference sequence
<reads_1.fastq>, <reads_2.fastq> Your input reads. In this case, mates of a paired end library
<output.sam> The name of your output file
Put it all together:
bwa mem -M -R '@RG\tID:sample_1\tLB:sample_1\tPL:ILLUMINA\tPM:HISEQ\tSM:sample_1' GCF_000001405.33_GRCh38.p7_chr20_genomic.fna read_1.fastq read_2.fastq > aligned_reads.sam
More information about this command in the BWA manual: http://bio-bwa.sourceforge.net/bwa.shtml
If everything worked, you should have a new aligned_reads.sam file. Doing a head command on this file shows us the beginning (the head) of this file, where we can see information such as the reference sequence name, read group info, the exact command that was used to produce this file, and the fist few aligned reads.
head aligned_reads.sam
@SQ SN:NC_000020.11 LN:64444167
@RG ID:sample_1 LB:sample_1 PL:ILLUMINA PM:HISEQ SM:sample_1
@PG ID:bwa PN:bwa VN:0.7.15-r1140 CL:bwa mem -M -R @RG\tID:sample_1\tLB:sample_1\tPL:ILLUMINA\tPM:HISEQ\tSM:sample_1 GCF_000001405.33_GRCh38.p7_chr20_genomic.fna read_1.fastq read_2.fastq
HS2000-940_146:5:1101:1161:63226 73 NC_000020.11 23775298 60 78M22S = 23775298 0 CTGNTAGCCCTGCTGAATCTCCCTCCTGACCCAACTCCCTCNTNNNNNNNGCTGGGTGACTGCTGNCNNCACNGGCTGTGNNNNNNNNNNNNNCAGCTGG ?@@#4ADDDFDFFHIGGFCFHCHFGIHGCGHEHHEHD3?BH#0#######--5CEECG=?AEEHE################################### NM:i:13 MD:Z:3G37C1C0T0A0C0T0C0T15T1C0T3T5 AS:i:52 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1161:63226 133 NC_000020.11 23775298 0 * = 23775298 0 NNCTCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGNNNNCNAAAGGAGCCTGGGT #################################################################################################### AS:i:0 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1262:12434 99 NC_000020.11 23843774 60 100M = 23843977 258 ATCAATGGTGTTTCTTTGCCAAGCTTCCTTAGTCGCCTTTAATCGGGAAAAGGTCTTCATTCTTTCTTGTCTTTGTTACCCTGTCATTTTTGAAGATAAC ?@@BDDFFFFHHHGHIIIHJEGHIIIGIJICHIGIIGIJIDHHGJJJ:;8EFH=CFHGGHIIIJJHHGBEHFFFFEEDCCCCEDCCADDEDD(5>5>@5@ NM:i:0 MD:Z:100 AS:i:100 XS:i:60 RG:Z:sample_1
HS2000-940_146:5:1101:1262:12434 147 NC_000020.11 23843977 60 55M45S = 23843774 -258 GTACATCATTCTGGGAGGCCAGGATACCATTGTCCAATTGCNNNNGGATGTTAATNNNNNNNNNNNNNNNNNNNNANNNNCTTCCNNNNNNNNCCACTCT #CDDCCC@38C<DDDDBBCDC;5ADDDCCACAADCC?=5,,####>CGHDC;;--####################1####FC<24########FDDD=B= NM:i:4 MD:Z:41T0G0A0T10 AS:i:47 XS:i:28 RG:Z:sample_1
HS2000-940_146:5:1101:1295:90112 83 NC_000020.11 23920564 60 100M = 23920284 -380 GCAAGTGAATGCTCTTTCCCACAGCAAAGGATTAACTGATTTCTGCTACTTGTGGCTCAGAGGCCAGGGACACTTGACCTGTCCTAGGAAGGCTGTCACC #@DDDD@><DDDEEAC?FFFHHEC=HECEAIGGE@IIGGHB;HAFD??F?IIJIEGGCIIJJJIHCIIIIIHIIHECIJIIHIJJJIFGHHFFDFFF@C@ NM:i:0 MD:Z:100 AS:i:100 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1295:90112 163 NC_000020.11 23920284 60 100M = 23920564 380 TGCCTTTCTNNACCGCCCACACTGTCACCCACNNNNNNTACACACTGGCTGGCAGTGGCCCCGGGAGCCAGGCCTCCCTGATAACTTGTTTTCTCTAAGT @@@FFFFFH##2<AAGHGDHHIGGIGGIJEEH######00?BB8AFHHIGCHGEE?=@DEDC>BB'83<98?12@?BBCDCD:>4:444:9ACCAA>>@C NM:i:9 MD:Z:9G0A21A0A0G0G0C0A27C34 AS:i:79 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1357:81190 83 NC_000020.11 23913876 60 100M = 23913621 -355 TGATGTGAATTCCTAACAGGAATTGGTGTAGGGGGCGGGCATGTGAACCGGCCCTGACCAATGAGACAGCAGGGGTGGTGAGCTGGGGAGTGGGTAGGAA 4CDDDDCCC>>?@CCCCCCCCABACCCDDDDDDBCAEDEC>GHEHEGJJIJJIIGIEIJJJJJJJGJIJJJJIJJJIJHFIJJJJJJHHHHHFFFFFCCC NM:i:0 MD:Z:100 AS:i:100 XS:i:44 RG:Z:sample_1
2) Sort sam and convert to bam
The algorithms used in downstream steps require the data to be sorted by coordinate and in bam format in order to be processed. We use Picard Tools and issue a single command to both sort the sam file produced in step 1 and output the resulting sorted data in bam format:
java -jar picard.jar SortSam INPUT=aligned_reads.sam OUTPUT=sorted_reads.bam SORT_ORDER=coordinate
More information about this command in the Picard Tools manual:
https://broadinstitute.github.io/picard/command-line-overview.html#SortSam
If this executed correctly, you should see something like the following:
[Wed Dec 07 11:38:40 EST 2016] picard.sam.SortSam INPUT=aligned_reads.sam OUTPUT=sorted_reads.bam SORT_ORDER=coordinate VERBOSITY=INFO QUIET=false VALIDATION_STRINGENCY=STRICT COMPRESSION_LEVEL=5 MAX_RECORDS_IN_RAM=500000 CREATE_INDEX=false CREATE_MD5_FILE=false
[Wed Dec 07 11:38:40 EST 2016] Executing as mk5636@compute-0-0.local on Linux 2.6.32-431.29.2.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0-b132; Picard version: 1.129(b508b2885562a4e932d3a3a60b8ea283b7ec78e2_1424706677) IntelDeflater
INFO 2016-12-07 11:39:05 SortSam Finished reading inputs, merging and writing to output now.
[Wed Dec 07 11:39:23 EST 2016] picard.sam.SortSam done. Elapsed time: 0.72 minutes.
Runtime.totalMemory()=1988100096
Let's take a look at the files before and after this step to see what happened. We will use samtools to view the sam/bam files.
Let's take a look at the first few lines of the original file. We'll use the samtools view command to view the sam file, and pipe the output to head -5 to show us only the 'head' of the file (in this case, the first 5 lines).
samtools view aligned_reads.sam | head -5
More information about samtools in the manual: http://www.htslib.org/doc/samtools.html
Output:
HS2000-940_146:5:1101:1161:63226 73 NC_000020.11 23775298 60 78M22S = 23775298 0 CTGNTAGCCCTGCTGAATCTCCCTCCTGACCCAACTCCCTCNTNNNNNNNGCTGGGTGACTGCTGNCNNCACNGGCTGTGNNNNNNNNNNNNNCAGCTGG ?@@#4ADDDFDFFHIGGFCFHCHFGIHGCGHEHHEHD3?BH#0#######--5CEECG=?AEEHE################################### NM:i:13 MD:Z:3G37C1C0T0A0C0T0C0T15T1C0T3T5 AS:i:52 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1161:63226 133 NC_000020.11 23775298 0 * = 23775298 0 NNCTCNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNNGNNNNCNAAAGGAGCCTGGGT #################################################################################################### AS:i:0 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1262:12434 99 NC_000020.11 23843774 60 100M = 23843977 258 ATCAATGGTGTTTCTTTGCCAAGCTTCCTTAGTCGCCTTTAATCGGGAAAAGGTCTTCATTCTTTCTTGTCTTTGTTACCCTGTCATTTTTGAAGATAAC ?@@BDDFFFFHHHGHIIIHJEGHIIIGIJICHIGIIGIJIDHHGJJJ:;8EFH=CFHGGHIIIJJHHGBEHFFFFEEDCCCCEDCCADDEDD(5>5>@5@ NM:i:0 MD:Z:100 AS:i:100 XS:i:0 RG:Z:sample_1
HS2000-940_146:5:1101:1262:12434 147 NC_000020.11 23843977 60 55M45S = 23843774 -258 GTACATCATTCTGGGAGGCCAGGATACCATTGTCCAATTGCNNNNGGATGTTAATNNNNNNNNNNNNNNNNNNNNANNNNCTTCCNNNNNNNNCCACTCT #CDDCCC@38C<DDDDBBCDC;5ADDDCCACAADCC?=5,,####>CGHDC;;--####################1####FC<24########FDDD=B= NM:i:4 MD:Z:41T0G0A0T10 AS:i:47 XS:i:28 RG:Z:sample_1
HS2000-940_146:5:1101:1295:90112 83 NC_000020.11 23920564 60 100M = 23920284 -380 GCAAGTGAATGCTCTTTCCCACAGCAAAGGATTAACTGATTTCTGCTACTTGTGGCTCAGAGGCCAGGGACACTTGACCTGTCCTAGGAAGGCTGTCACC #@DDDD@><DDDEEAC?FFFHHEC=HECEAIGGE@IIGGHB;HAFD??F?IIJIEGGCIIJJJIHCIIIIIHIIHECIJIIHIJJJIFGHHFFDFFF@C@ NM:i:0 MD:Z:100 AS:i:100 XS:i:0 RG:Z:sample_1
Let's compare this initial alignment file to the new sorted file:
samtools view sorted_reads.bam | head -5
Output:
HS2000-940_146:5:2109:14063:29918 161 NC_000020.11 64145 4 54S46M = 23724989 23660944 TTCCAATCCATTCCATTCCATCACACTGCATTCCATTCCATTCCAATCCCCTCAACTCCACTCCACTCCACTCCATTCCACTCCAATCAATTCCATTGCA @CCFFFFFHGDHHJIJJJJJIJIFHHGCGIHHIJJJGHIHIIIJJIHIGGGHIJJE:FFHIGIDHJGIGGIJJ@;CDHGGEIHHHEHF;CCB>;;3;>;> MD:Z:6G27C8T2 RG:Z:sample_1 NM:i:3 AS:i:33 XS:i:31
HS2000-940_146:5:2110:1521:37886 163 NC_000020.11 1217420 0 55S21M24S = 1217591 271 GACAGTTCTGAAGAGAGCAGGGGTTCTTCCAGCATTGCATTTGAGCTCCGAAAATGGACAGACTGCCTCCTCAAGTCGGTCCTTGACCTCCGTGCACCCT ?7:DDD:B,+ADD43C?BF++<2<):**11*1:;C*0?0B?F>GCDBF30'-'-8@8..@1@E@;37@)?76@########################### MD:Z:21 RG:Z:sample_1 NM:i:0 AS:i:21 XS:i:34
HS2000-940_146:5:2110:1521:37886 83 NC_000020.11 1217591 0 100M = 1217420 -271 ATATCCAGACAAACAGGGTCTGGAGTAGACCTCCAGCAAATTCCAACAGACCTGCAGCTGAGGGTCCTGACTGTTAGAAGGAAAACTAACAAACAGAAAG CA@>9CA;5?@>6;;..1;77;3=77)75CCF=)>HG@>BB3GEIGFB??BHDIIIGBHGFC:13@ACF9CAA,@F?EDA4IGGGAGHHHGHEFFFD?@@ MD:Z:3C4G17G13C59 RG:Z:sample_1 NM:i:4 AS:i:81 XS:i:81
HS2000-940_146:5:1102:10582:53061 113 NC_000020.11 1544082 22 100M = 23852837 22308739 TTCTGTTGATTTGGGGTGGAGAGTTCTGTAGAGGTCTGTTAGGTCTGCTTGGTCCAGAGCTGAGCTCAAATCCTGAATATCCTTGTTAATTTTCTGTCTC ###CCA:(>(;?:8;A>A>D@>D@=3=>7=@72@@AED@7BFCF?EIIF?0;IGAFD9HFD9D9@>E?9+<HCIGHEA?C3DDFDGBFD?C?DDAAD?;; MD:Z:32T4A26T4G30 RG:Z:sample_1 NM:i:4 AS:i:80 XS:i:70
HS2000-940_146:5:1112:8371:47601 99 NC_000020.11 2502086 10 100M = 2502334 348 AACTAGAATAACCAATGCAGAGAAGTCCTTAAAGGACCTGATGGAGCTGAAAACCAAGGCACAAGAACTACGTGATGAATACACAAGCCTCAGTAGCCGA CCCFFFFFHHHHHIJIIJJJJGJJIHHIIJJJJIJGIJJIGGHGHHGGIIJJJJJJJGHGGGIIJIIGHHHGEDFEEFEEEEEEDDDBDDDCC>CCDDBB MD:Z:33A22T5G12C4G19 RG:Z:sample_1 NM:i:5 AS:i:75 XS:i:79
Is the output consistent with what we expect?
3) Alignment Metrics
Let's compute some statistics to see how well our reads aligned to the reference genome. We'll use samtools flagstat for this.
samtools flagstat aligned_reads.sam
Output:
194492 + 0 in total (QC-passed reads + QC-failed reads)
80 + 0 secondary
0 + 0 supplementary
0 + 0 duplicates
193804 + 0 mapped (99.65% : N/A)
194412 + 0 paired in sequencing
97206 + 0 read1
97206 + 0 read2
190812 + 0 properly paired (98.15% : N/A)
193108 + 0 with itself and mate mapped
616 + 0 singletons (0.32% : N/A)
0 + 0 with mate mapped to a different chr
0 + 0 with mate mapped to a different chr (mapQ>=5)
Hint: Save these metrics to a text file by redirecting the output to a new file
samtools flagstat aligned_reads.sam > alignment_metrics.txt
4) Mark Duplicates
During the sequencing process, the same DNA fragments may be sequenced several times. These duplicate reads are not informative and cannot be considered as evidence for or against a putative variant. For example, duplicates can arise during sample preparation e.g. library construction using PCR. Without this step, you risk having over-representation in your sequence of areas preferentially amplified during PCR. Duplicate reads can also result from a single amplification cluster, incorrectly detected as multiple clusters by the optical sensor of the sequencing instrument. These duplication artifacts are referred to as optical duplicates.
We use Picard Tools to locate and tag duplicate reads in a BAM or SAM file, where duplicate reads are defined as originating from a single fragment of DNA.
Note that this step does not remove the duplicate reads, but rather flags them as such in the read's SAM record. We'll take a look at how this is done shortly. Downstream GATK tools will ignore reads flagged as duplicates by default.
Note: Duplicate marking should not be applied to amplicon sequencing or other data types where reads start and stop at the same positions by design. To keep duplicate reads, skip this step and use the sorted_reads.bam file in place of dedupreads.bam going forward.
java -jar picard.jar MarkDuplicates INPUT=sorted_reads.bam OUTPUT=dedup_reads.bam METRICS_FILE=metrics.txt
If this executed correctly, you should see something like the following:
[Mon Dec 19 17:29:19 EST 2016] Executing as mk5636@compute-13-30.local on Linux 2.6.32-431.29.2.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.8.0-b132; Picard version: 1.129(b508b2885562a4e932d3a3a60b8ea283b7ec78e2_1424706677) IntelDeflater
INFO 2016-12-19 17:29:19 MarkDuplicates Start of doWork freeMemory: 797279896; totalMemory: 798490624; maxMemory: 11276386304
INFO 2016-12-19 17:29:19 MarkDuplicates Reading input file and constructing read end information.
INFO 2016-12-19 17:29:19 MarkDuplicates Will retain up to 43370716 data points before spilling to disk.
INFO 2016-12-19 17:29:22 MarkDuplicates Read 194420 records. 0 pairs never matched.
INFO 2016-12-19 17:29:22 MarkDuplicates After buildSortedReadEndLists freeMemory: 540268072; totalMemory: 909639680; maxMemory: 11276386304
INFO 2016-12-19 17:29:22 MarkDuplicates Will retain up to 352387072 duplicate indices before spilling to disk.
INFO 2016-12-19 17:29:23 MarkDuplicates Traversing read pair information and detecting duplicates.
INFO 2016-12-19 17:29:23 MarkDuplicates Traversing fragment information and detecting duplicates.
INFO 2016-12-19 17:29:23 MarkDuplicates Sorting list of duplicate records.
INFO 2016-12-19 17:29:24 MarkDuplicates After generateDuplicateIndexes freeMemory: 906398800; totalMemory: 3729260544; maxMemory: 11276386304
INFO 2016-12-19 17:29:24 MarkDuplicates Marking 15269 records as duplicates.
INFO 2016-12-19 17:29:24 MarkDuplicates Found 31 optical duplicate clusters.
INFO 2016-12-19 17:29:27 MarkDuplicates Before output close freeMemory: 3757499808; totalMemory: 3762290688; maxMemory: 11276386304
INFO 2016-12-19 17:29:27 MarkDuplicates After output close freeMemory: 3753828760; totalMemory: 3758620672; maxMemory: 11276386304
[Mon Dec 19 17:29:27 EST 2016] picard.sam.markduplicates.MarkDuplicates done. Elapsed time: 0.14 minutes.
Runtime.totalMemory()=3758620672
These stats are broken down in the metrics.txt file:
LIBRARY UNPAIRED_READS_EXAMINED READ_PAIRS_EXAMINED UNMAPPED_READS UNPAIREDD_READ_DUPLICATES READ_PAIR_DUPLICATE READ_PAIR_OPTICAL_DUPLICATES PERCENT_DUPLICATION ESTIMATED_LIBRARY_SIZE
sample_1 616 96554 688 165 7552 31 0.078818 586767
Let's take a look at the bam file before and after the Mark Duplicates step to see how reads are flagged as duplicates.
Refresher: The second column in a SAM file is known as the bitwise flag. This flag allows for the storage of lots of information in a highly efficient format. Let's look at the first read in sorted_reads.bam:
samtools view sorted_reads.bam | head -1
HS2000-940_146:5:2109:14063:29918 161 NC_000020.11 64145 4 54S46M = 23724989 23660944 TTCCAATCCATTCCATTCCATCACACTGCATTCCATTCCATTCCAATCCCCTCAACTCCACTCCACTCCACTCCATTCCACTCCAATCAATTCCATTGCA @CCFFFFFHGDHHJIJJJJJIJIFHHGCGIHHIJJJGHIHIIIJJIHIGGGHIJJE:FFHIGIDHJGIGGIJJ@;CDHGGEIHHHEHF;CCB>;;3;>;> MD:Z:6G27C8T2 RG:Z:sample_1 NM:i:3 AS:i:33 XS:i:31
Question: What is the bitwise flag value for this read?
(Answer: 161)
Question: What does this value represent? http://broadinstitute.github.io/picard/explain-flags.html
(Answer: read paired, mate reverse strand, second in pair)
Note: "read is PCR or optical duplicate" is also stored in this flag
Let's look at this read before and after marking duplicates: HS2000-1010_101:8:2205:14144:55120
samtools view sorted_reads.bam | grep 'HS2000-1010_101:8:2205:14144:55120'
HS2000-1010_101:8:2205:14144:55120 161 NC_000020.11 24013181 60 100M = 24013243 162 TACTGTCCTGTGTTTGTTCATTATTCCCCATGTTTCCTAAGATATGTTTTCTAAGCCAACACATTAGTTCAAATTACTGCATTTTTCTTGAATCTTGACA @@@DDDDDDFHFHHIDHEDHHGEJIIEGGGIGEEGHIIIIIJFGIEICFHFGIJGDDFADHEHBFHI;@F@GGFE@CDH@??ACA>@BDFD>?;A>6>C3 MD:Z:40T59 RG:Z:sample_1 NM:i:1 AS:i:95 XS:i:0
HS2000-1010_101:8:2205:14144:55120 81 NC_000020.11 24013243 60 100M = 24013181 -162 ATTAGTTCAAATTACTGCATTTTTCTTGAATCTTGACAAGAAAATTATGTAGGAAGTAGATTTGAGTTTTTGCGTAGCTGTGTCTACTGTGACCCAATGG CCCCCCC@>DC@CACECCCBB@>@ACHHEHDD@IHEIIIHIF>GEIIIHGHIGGIIGFBFACGHGGHEBGGHGGHFEDFCDIIIGHHFC?A:?BFFB@@? MD:Z:73A26 RG:Z:sample_1 NM:i:1 AS:i:95 XS:i:19
samtools view dedup_reads.bam | grep 'HS2000-1010_101:8:2205:14144:55120'
HS2000-1010_101:8:2205:14144:55120 1185 NC_000020.11 24013181 60 100M = 24013243 162 TACTGTCCTGTGTTTGTTCATTATTCCCCATGTTTCCTAAGATATGTTTTCTAAGCCAACACATTAGTTCAAATTACTGCATTTTTCTTGAATCTTGACA @@@DDDDDDFHFHHIDHEDHHGEJIIEGGGIGEEGHIIIIIJFGIEICFHFGIJGDDFADHEHBFHI;@F@GGFE@CDH@??ACA>@BDFD>?;A>6>C3 MD:Z:40T59 PG:Z:MarkDuplicates RG:Z:sample_1 NM:i:1 AS:i:95XS:i:0
HS2000-1010_101:8:2205:14144:55120 1105 NC_000020.11 24013243 60 100M = 24013181 -162 ATTAGTTCAAATTACTGCATTTTTCTTGAATCTTGACAAGAAAATTATGTAGGAAGTAGATTTGAGTTTTTGCGTAGCTGTGTCTACTGTGACCCAATGG CCCCCCC@>DC@CACECCCBB@>@ACHHEHDD@IHEIIIHIF>GEIIIHGHIGGIIGFBFACGHGGHEBGGHGGHFEDFCDIIIGHHFC?A:?BFFB@@? MD:Z:73A26 PG:Z:MarkDuplicates RG:Z:sample_1 NM:i:1 AS:i:95XS:i:19
5) Prepare reference dictionary, fasta index, and bam index
In order to run GATK, we need to build a reference dictionary, fasta index, and a bam index.
We use Picard Tools to build the reference dictionary for GATK:
java -jar picard.jar CreateSequenceDictionary R=GCF_000001405.33_GRCh38.p7_chr20_genomic.fna O=GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.dict
We use samtools to build the fasta index:
samtools faidx GCF_000001405.33_GRCh38.p7_chr20_genomic.fna
We use samtools to build the bam index:
samtools index dedup_reads.bam
We should have 3 new files:
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.dict - GATK reference dictionary
GCF_000001405.33_GRCh38.p7_chr20_genomic.fna.fai - fasta Index
dedup_reads.bam.bai - bam index
6) Base Quality Score Recalibration
Variant calling algorithms rely heavily on the quality score assigned to the individual base calls in each sequence read. This is because the quality score tells us how much we can trust that particular observation to inform us about the biological truth of the site where that base aligns. If we have a basecall that has a low quality score, that means we're not sure we actually read that A correctly, and it could actually be something else. So we won't trust it as much as other base calls that have higher qualities. In other words we use that score to weigh the evidence that we have for or against a variant allele existing at a particular site. [https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS]
Refresher: What are quality scores?
- Per-base estimates of error emitted by the sequencer
- Expresses the level of confidence for each base called
- Use standard Pred scores: Q20 is a general cutoff for high quality and represents 99% certainty that a base was called correctly
- 99% certainty means 1 out of 100 expected to be wrong. Let's consider a small dataset of 1M reads with a read length of 50, this means 50M bases. With 99% confidence, this means 50,000 possible erroneous bases.
The image below shows an example of average quality score at east position in the read, for all reads in a library (output from FastQC)
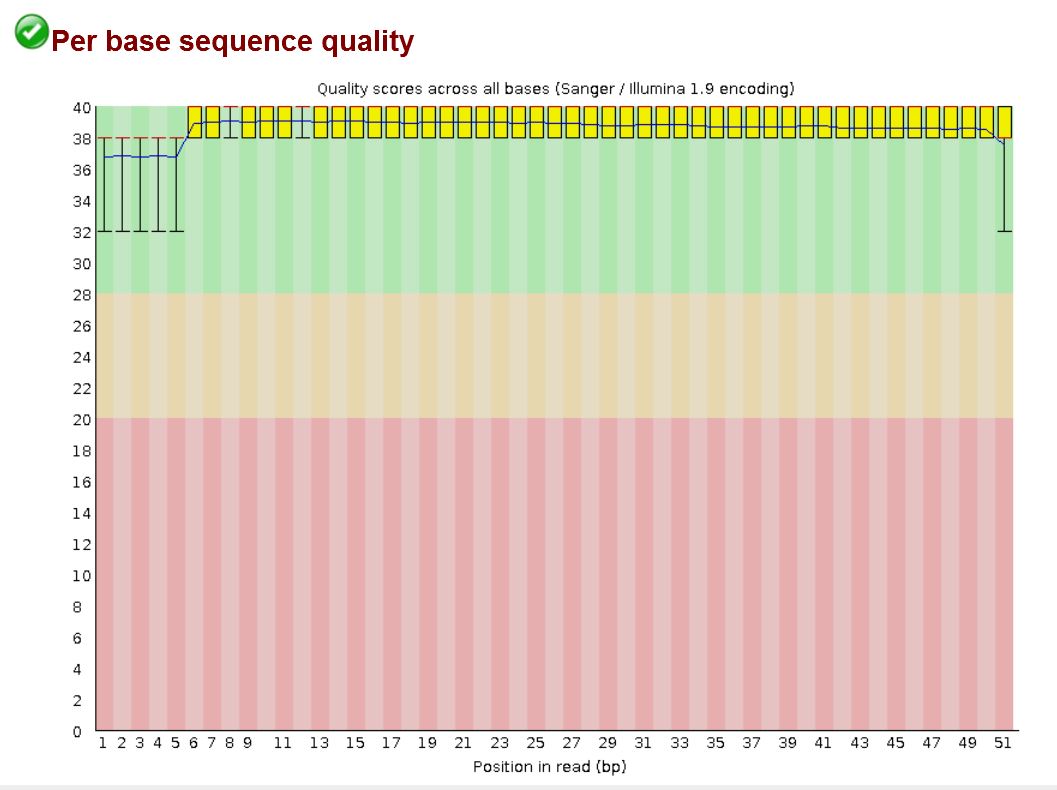
The image below shows individual quality scores (blue bars) for each position in a single read. The horizontal blue line represents the Q20 phred score value.

Quality scores emitted by sequencing machines are biased and inaccurate
Unfortunately the scores produced by the machines are subject to various sources of systematic technical error, leading to over- or under-estimated base quality scores in the data. Some of these errors are due to the physics or the chemistry of how the sequencing reaction works, and some are probably due to manufacturing flaws in the equipment. Base quality score recalibration (BQSR) is a process in which we apply machine learning to model these errors empirically and adjust the quality scores accordingly. This allows us to get more accurate base qualities, which in turn improves the accuracy of our variant calls. [https://software.broadinstitute.org/gatk/best-practices/bp_3step.php?case=GermShortWGS]
How BQSR works
You provide GATK Base Recalibrator with a set of known variants.
GATK Base Recalibrator analyzes all reads looking for mismatches between the read and reference, skipping those positions which are included in the set of known variants (from step 1).
GATK Base Recalibrator computes statistics on the mismatches (identified in step 2) based on the reported quality score, the position in the read, the sequencing context (ex: preceding and current nucleotide).
Based on the statistics computed in step 3, an empirical quality score is assigned to each mismatch, overwriting the original reported quality score.
As an example, pre-calibration a file could contain only reported Q25 bases, which seems good. However, it may be that these bases actually mismatch the reference at a 1 in 100 rate, so are actually Q20. These higher-than-empirical quality scores provide false confidence in the base calls. Moreover, as is common with sequencing-by-synthesis machines, base mismatches with the reference occur at the end of the reads more frequently than at the beginning. Also, mismatches are strongly associated with sequencing context, in that the dinucleotide AC is often much lower quality than TG.
[http://gatkforums.broadinstitute.org/gatk/discussion/44/base-quality-score-recalibration-bqsr]
Note that this step requires a 'truth' or 'known' set of variants. For this example we will be using the gold set from the 1000 genomes project (provided in the sample dataset: 1000G_omni2.5.hg38.vcf.gz.tbi). An index for the VCF is required as well and is also provided. If you need to build an index for your VCF file, you can build one easily using the TABIX program, like so:
tabix -p vcf 1000G_omni2.5.hg38.vcf.gz
More information about the tabix command in the manual: http://www.htslib.org/doc/tabix.html
Step 1: Analyze Covaration
java -jar GenomeAnalysisTK.jar -T BaseRecalibrator -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I dedup_reads.bam -knownSites 1000G_omni2.5.hg38.vcf.gz -o recal_data.table
Learn more about this command in the GATK documentation: https://software.broadinstitute.org/gatk/gatkdocs/3.5-0/
If executed correctly, you should see something like this:
INFO 13:53:12,940 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:53:12,966 HelpFormatter - The Genome Analysis Toolkit (GATK) v3.5-0-g36282e4, Compiled 2015/11/25 04:03:56
INFO 13:53:12,966 HelpFormatter - Copyright (c) 2010 The Broad Institute
INFO 13:53:12,966 HelpFormatter - For support and documentation go to http://www.broadinstitute.org/gatk
INFO 13:53:12,970 HelpFormatter - Program Args: -T BaseRecalibrator -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I dedup_reads.bam -knownSites 1000G_omni2.5.hg38.vcf.gz -o recal_data.table
INFO 13:53:12,977 HelpFormatter - Executing as mk5636@compute-21-2.local on Linux 3.10.0-327.10.1.el7.x86_64 amd64; OpenJDK 64-Bit Server VM 1.8.0_92-b15.
INFO 13:53:12,978 HelpFormatter - Date/Time: 2017/01/12 13:53:12
INFO 13:53:12,978 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:53:12,978 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:53:13,179 GenomeAnalysisEngine - Strictness is SILENT
INFO 13:53:13,263 GenomeAnalysisEngine - Downsampling Settings: No downsampling
INFO 13:53:13,268 SAMDataSource$SAMReaders - Initializing SAMRecords in serial
INFO 13:53:13,313 SAMDataSource$SAMReaders - Done initializing BAM readers: total time 0.02
WARN 13:53:13,717 IndexDictionaryUtils - Track knownSites doesn't have a sequence dictionary built in, skipping dictionary validation
INFO 13:53:14,139 GenomeAnalysisEngine - Preparing for traversal over 1 BAM files
INFO 13:53:14,143 GenomeAnalysisEngine - Done preparing for traversal
INFO 13:53:14,143 ProgressMeter - [INITIALIZATION COMPLETE; STARTING PROCESSING]
INFO 13:53:14,144 ProgressMeter - | processed | time | per 1M | | total | remaining
INFO 13:53:14,144 ProgressMeter - Location | reads | elapsed | reads | completed | runtime | runtime
INFO 13:53:14,250 BaseRecalibrator - The covariates being used here:
INFO 13:53:14,250 BaseRecalibrator - ReadGroupCovariate
INFO 13:53:14,250 BaseRecalibrator - QualityScoreCovariate
INFO 13:53:14,250 BaseRecalibrator - ContextCovariate
INFO 13:53:14,251 ContextCovariate - Context sizes: base substitution model 2, indel substitution model 3
INFO 13:53:14,251 BaseRecalibrator - CycleCovariate
INFO 13:53:14,254 ReadShardBalancer$1 - Loading BAM index data
INFO 13:53:14,255 ReadShardBalancer$1 - Done loading BAM index data
INFO 13:53:31,295 BaseRecalibrator - Calculating quantized quality scores...
INFO 13:53:31,325 BaseRecalibrator - Writing recalibration report...
INFO 13:53:33,902 BaseRecalibrator - ...done!
INFO 13:53:33,902 BaseRecalibrator - BaseRecalibrator was able to recalibrate 178205 reads
INFO 13:53:33,904 ProgressMeter - done 178205.0 19.0 s 110.0 s 99.3% 19.0 s 0.0 s
INFO 13:53:33,904 ProgressMeter - Total runtime 19.76 secs, 0.33 min, 0.01 hours
INFO 13:53:33,904 MicroScheduler - 16278 reads were filtered out during the traversal out of approximately 194483 total reads (8.37%)
INFO 13:53:33,905 MicroScheduler - -> 0 reads (0.00% of total) failing BadCigarFilter
INFO 13:53:33,905 MicroScheduler - -> 15258 reads (7.85% of total) failing DuplicateReadFilter
INFO 13:53:33,905 MicroScheduler - -> 0 reads (0.00% of total) failing FailsVendorQualityCheckFilter
INFO 13:53:33,905 MicroScheduler - -> 0 reads (0.00% of total) failing MalformedReadFilter
INFO 13:53:33,905 MicroScheduler - -> 0 reads (0.00% of total) failing MappingQualityUnavailableFilter
INFO 13:53:33,906 MicroScheduler - -> 985 reads (0.51% of total) failing MappingQualityZeroFilter
INFO 13:53:33,906 MicroScheduler - -> 35 reads (0.02% of total) failing NotPrimaryAlignmentFilter
INFO 13:53:33,906 MicroScheduler - -> 0 reads (0.00% of total) failing UnmappedReadFilter
Step 2: Apply BQSR
This step applies the recalibration computed in the Step 1 to the bam file.
java -jar GenomeAnalysisTK.jar -T PrintReads -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I dedup_reads.bam -BQSR recal_data.table -o recal_reads.bam
If everything worked, you should see something like this:
INFO 13:58:17,934 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:58:17,936 HelpFormatter - The Genome Analysis Toolkit (GATK) v3.5-0-g36282e4, Compiled 2015/11/25 04:03:56
INFO 13:58:17,937 HelpFormatter - Copyright (c) 2010 The Broad Institute
INFO 13:58:17,937 HelpFormatter - For support and documentation go to http://www.broadinstitute.org/gatk
INFO 13:58:17,940 HelpFormatter - Program Args: -T PrintReads -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I dedup_reads.bam -BQSR recal_data.table -o recal_reads.bam
INFO 13:58:17,947 HelpFormatter - Executing as mk5636@compute-21-2.local on Linux 3.10.0-327.10.1.el7.x86_64 amd64; OpenJDK 64-Bit Server VM 1.8.0_92-b15.
INFO 13:58:17,947 HelpFormatter - Date/Time: 2017/01/12 13:58:17
INFO 13:58:17,948 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:58:17,948 HelpFormatter - --------------------------------------------------------------------------------
INFO 13:58:18,051 GenomeAnalysisEngine - Strictness is SILENT
INFO 13:58:18,648 ContextCovariate - Context sizes: base substitution model 2, indel substitution model 3
INFO 13:58:18,683 GenomeAnalysisEngine - Downsampling Settings: No downsampling
INFO 13:58:18,689 SAMDataSource$SAMReaders - Initializing SAMRecords in serial
INFO 13:58:18,713 SAMDataSource$SAMReaders - Done initializing BAM readers: total time 0.02
INFO 13:58:18,791 GenomeAnalysisEngine - Preparing for traversal over 1 BAM files
INFO 13:58:18,794 GenomeAnalysisEngine - Done preparing for traversal
INFO 13:58:18,795 ProgressMeter - [INITIALIZATION COMPLETE; STARTING PROCESSING]
INFO 13:58:18,795 ProgressMeter - | processed | time | per 1M | | total | remaining
INFO 13:58:18,795 ProgressMeter - Location | reads | elapsed | reads | completed | runtime | runtime
INFO 13:58:18,800 ReadShardBalancer$1 - Loading BAM index data
INFO 13:58:18,801 ReadShardBalancer$1 - Done loading BAM index data
INFO 13:58:40,267 Walker - [REDUCE RESULT] Traversal result is: org.broadinstitute.gatk.engine.io.stubs.SAMFileWriterStub@42238078
INFO 13:58:40,378 ProgressMeter - done 194483.0 21.0 s 110.0 s 99.0% 21.0 s 0.0 s
INFO 13:58:40,379 ProgressMeter - Total runtime 21.58 secs, 0.36 min, 0.01 hours
INFO 13:58:40,379 MicroScheduler - 0 reads were filtered out during the traversal out of approximately 194483 total reads (0.00%)
INFO 13:58:40,379 MicroScheduler - -> 0 reads (0.00% of total) failing BadCigarFilter
INFO 13:58:40,379 MicroScheduler - -> 0 reads (0.00% of total) failing MalformedReadFilter
The output of this step, recal_reads.bam, is our analysis-ready dataset that we will provide to the variant calling tool in the next step of the analysis.
Supplementary material: What to do if you don't have a set of known variants?
BQSR is an optional but highly recommended step in variant calling analysis. In the event you are working with an organism for which there is no known set of variants available, it is possible to produce a set of known variants for use in this step, although it does require some additional processing steps.
This procedure is known as bootstrapping and entails calling variants without running BQSR, filtering those variants to obtain a high confidence set of variants, and then using these variants as input for the BQSR step. This process can be repeated until convergence.