Variant Discovery
Once you have a pre-processed, analysis-ready bam file, you can begin the variant discovery process.
Overview
Call variants
Filter variants
Annotation
Visualization
1) Call Variants
We use the GATK HaplotypeCaller to perform variant calling. The HaplotypeCaller is capable of calling SNPs and indels simultaneously via local de-novo assembly of haplotypes in an active region. In other words, whenever the program encounters a region showing signs of variation, it discards the existing mapping information and completely reassembles the reads in that region. This step is designed to maximize sensitivity in order to minimize false negatives, i.e. failing to identify real variants.
java -jar GenomeAnalysisTK.jar -T HaplotypeCaller -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I recal_reads.bam -o raw_variants.vcf
If everything worked you should see something like the following:
INFO 18:21:22,387 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:21:22,389 HelpFormatter - The Genome Analysis Toolkit (GATK) v3.3-0-g37228af, Compiled 2014/10/24 01:07:22
INFO 18:21:22,389 HelpFormatter - Copyright (c) 2010 The Broad Institute
INFO 18:21:22,389 HelpFormatter - For support and documentation go to http://www.broadinstitute.org/gatk
INFO 18:21:22,392 HelpFormatter - Program Args: -T HaplotypeCaller -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -I recal_reads.bam -o raw_variants.vcf
INFO 18:21:22,396 HelpFormatter - Executing as mk5636@compute-0-5.local on Linux 2.6.32-431.29.2.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.7.0_60-b19.
INFO 18:21:22,396 HelpFormatter - Date/Time: 2016/12/20 18:21:22
INFO 18:21:22,396 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:21:22,397 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:21:22,790 GenomeAnalysisEngine - Strictness is SILENT
INFO 18:21:22,834 GenomeAnalysisEngine - Downsampling Settings: Method: BY_SAMPLE, Target Coverage: 250
INFO 18:21:22,840 SAMDataSource$SAMReaders - Initializing SAMRecords in serial
INFO 18:21:22,850 SAMDataSource$SAMReaders - Done initializing BAM readers: total time 0.01
INFO 18:21:22,856 HCMappingQualityFilter - Filtering out reads with MAPQ < 20
INFO 18:21:22,908 GenomeAnalysisEngine - Preparing for traversal over 1 BAM files
INFO 18:21:22,973 GenomeAnalysisEngine - Done preparing for traversal
INFO 18:21:22,973 ProgressMeter - [INITIALIZATION COMPLETE; STARTING PROCESSING]
INFO 18:21:22,974 ProgressMeter - | processed | time | per 1M | | total | remaining
INFO 18:21:22,974 ProgressMeter - Location | active regions | elapsed | active regions | completed | runtime | runtime
INFO 18:21:22,974 HaplotypeCaller - Disabling physical phasing, which is supported only for reference-model confidence output
INFO 18:21:23,055 HaplotypeCaller - Using global mismapping rate of 45 => -4.5 in log10 likelihood units
Using AVX accelerated implementation of PairHMM
INFO 18:21:31,094 VectorLoglessPairHMM - libVectorLoglessPairHMM unpacked successfully from GATK jar file
INFO 18:21:31,094 VectorLoglessPairHMM - Using vectorized implementation of PairHMM
INFO 18:21:52,976 ProgressMeter - NC_000020.11:23850907 0.0 30.0 s 49.6 w 37.0% 81.0 s 51.0 s
Time spent in setup for JNI call : 0.023440074
Total compute time in PairHMM computeLikelihoods() : 3.811236998
INFO 18:22:12,892 HaplotypeCaller - Ran local assembly on 1062 active regions
INFO 18:22:13,090 ProgressMeter - done 6.4444167E7 50.0 s 0.0 s 100.0% 50.0 s 0.0 s
INFO 18:22:13,090 ProgressMeter - Total runtime 50.12 secs, 0.84 min, 0.01 hours
INFO 18:22:13,090 MicroScheduler - 16568 reads were filtered out during the traversal out of approximately 194420 total reads (8.52%)
INFO 18:22:13,090 MicroScheduler - -> 15234 reads (7.84% of total) failing DuplicateReadFilter
INFO 18:22:13,091 MicroScheduler - -> 0 reads (0.00% of total) failing FailsVendorQualityCheckFilter
INFO 18:22:13,091 MicroScheduler - -> 1300 reads (0.67% of total) failing HCMappingQualityFilter
INFO 18:22:13,091 MicroScheduler - -> 0 reads (0.00% of total) failing MalformedReadFilter
INFO 18:22:13,091 MicroScheduler - -> 0 reads (0.00% of total) failing MappingQualityUnavailableFilter
INFO 18:22:13,091 MicroScheduler - -> 34 reads (0.02% of total) failing NotPrimaryAlignmentFilter
INFO 18:22:13,091 MicroScheduler - -> 0 reads (0.00% of total) failing UnmappedReadFilter
INFO 18:22:13,933 GATKRunReport - Uploaded run statistics report to AWS S3
And have two new output files:
raw_variants.vcf - all the sites that the HaplotypeCaller evaluated to be potentially variant. Note that this file contains both SNPs and Indels.
raw_variants.vcf.idx - index file
Extract SNPs and Indels
This step separates SNPs and Indels so they can be processed and analyzed independently.
java -jar GenomeAnalysisTK.jar -T SelectVariants -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -V raw_variants.vcf -selectType SNP -o raw_snps.vcf
java -jar GenomeAnalysisTK.jar -T SelectVariants -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -V raw_variants.vcf -selectType INDEL -o raw_indels.vcf
You should see something like this for each command:
INFO 18:38:11,974 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:38:11,976 HelpFormatter - The Genome Analysis Toolkit (GATK) v3.3-0-g37228af, Compiled 2014/10/24 01:07:22
INFO 18:38:11,976 HelpFormatter - Copyright (c) 2010 The Broad Institute
INFO 18:38:11,976 HelpFormatter - For support and documentation go to http://www.broadinstitute.org/gatk
INFO 18:38:11,979 HelpFormatter - Program Args: -T SelectVariants -R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna -V raw_variants.vcf -selectType SNP -o raw_snps.vcf
INFO 18:38:11,983 HelpFormatter - Executing as mk5636@compute-0-5.local on Linux 2.6.32-431.29.2.el6.x86_64 amd64; Java HotSpot(TM) 64-Bit Server VM 1.7.0_60-b19.
INFO 18:38:11,983 HelpFormatter - Date/Time: 2016/12/20 18:38:11
INFO 18:38:11,984 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:38:11,984 HelpFormatter - --------------------------------------------------------------------------------
INFO 18:38:12,372 GenomeAnalysisEngine - Strictness is SILENT
INFO 18:38:12,419 GenomeAnalysisEngine - Downsampling Settings: Method: BY_SAMPLE, Target Coverage: 1000
INFO 18:38:12,508 GenomeAnalysisEngine - Preparing for traversal
INFO 18:38:12,509 GenomeAnalysisEngine - Done preparing for traversal
INFO 18:38:12,509 ProgressMeter - [INITIALIZATION COMPLETE; STARTING PROCESSING]
INFO 18:38:12,510 ProgressMeter - | processed | time | per 1M | | total | remaining
INFO 18:38:12,510 ProgressMeter - Location | sites | elapsed | sites | completed | runtime | runtime
INFO 18:38:13,264 SelectVariants - 1560 records processed.
INFO 18:38:13,480 ProgressMeter - done 1932.0 0.0 s 8.4 m 67.4% 0.0 s 0.0 s
INFO 18:38:13,481 ProgressMeter - Total runtime 0.97 secs, 0.02 min, 0.00 hours
INFO 18:38:13,873 GATKRunReport - Uploaded run statistics report to AWS S3
And have four new output files in total after running both commands:
raw_snps.vcf and raw_indels.vcf - VCF files containing only snps and only indels
raw_snps.vcf.idx and raw_indels.vcf.idx- index files
2) Filter Variants
The first step is designed to maximize sensitivity and is thus very lenient in calling variants. This is good because it minimizes the chance of missing real variants, but it means that we need to filter the raw callset produced in order to reduce the amount of false positives, which can be quite large. This is an important step in order to obtain the the highest-quality call set possible.
We apply the recommended hard filters for SNPs and Indels.
SNPs
java -jar GenomeAnalysisTK.jar -T VariantFiltration \
-R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna \
-V raw_snps.vcf \
-filterName "QD_filter" \
-filter "QD'<'2.0" \
-filterName "FS_filter" \
-filter "FS'>'60.0" \
-filterName "MQ_filter" \
-filter "MQ'<'40.0" \
-filterName "SOR_filter" \
-filter "SOR'>'4.0" \
-o filtered_snps.vcf
You should get two new files: filtered_snps.vcfand filtered_snps.vcf.idx
Note: SNPs which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however, they will be marked as ‘*_filter’ based on which filter the SNP failed, while SNPs which passed the filter will be marked as ‘PASS’
Indels
java -jar GenomeAnalysisTK.jar -T VariantFiltration \
-R GCF_000001405.33_GRCh38.p7_chr20_genomic.fna \
-V raw_indels.vcf \
-filterName "QD_filter" \
-filter "QD'<'2.0" \
-filterName "FS_filter" \
-filter "FS'>'200.0" \
-filterName "SOR_filter" \
-filter "SOR'>'10.0" \
-o filtered_indels.vcf
You should get two new files: filtered_indels.vcfand filtered_indels.vcf.idx
Note: Indels which are ‘filtered out’ at this step will remain in the filtered_snps.vcf file, however, they will be marked as ‘*_filter’ based on which filter the indel failed, while Indels which passed the filter will be marked as ‘PASS’
3) Annotation
We will use the program SnpEff. It annotates and predicts the effects of variants on genes (such as amino acid changes).
SnpEff has pre-built databases for thousands of genomes. We will be using the pre-built database GRCh38.p2.RefSeq.
Caveat:
Although we are using the NCBI RefSeq SnpEff Database, there is still an incompatibility in the chromosome names (ie. in our data, chromosome 20 is named NC_000020.11 whereas in the prebuilt SnpEff database, chromosome 20 is named '20'). To get around this, we will do a little magic to transform 'NC_000020.11' to '20'.
First let's make a copy of filtered_snps.vcf:
cp filtered_snps.vcf filtered_snps_renamed.vcf
We will use vim to edit our new file:
vi filtered_snps_renamed.vcf
Use this snippet to replace all instanced of 'NC_000020.11' with '20':
:%s/NC_000020.11/20/g
Save changes and quit:
:wq <return>
We're now ready to run SnpEff:
java -jar /scratch/gencore/software/snpEff/4.1/snpEff.jar -v GRCh38.p2.RefSeq filtered_snps_renamed.vcf > filtered_snps.ann.vcf
Upon completion, you should see three new output files:
filtered_snps.ann.vcf - An annotated VCF filesnpEff_genes.txt - A tab-delimited file summarizing the genes found to contain variantssnpEff_summary.html - An HTML summary report
Locating and downloading the SnpEff database for your organism:
To locate and download the SnpEff database for your organism, execute the following command:
java -jar /share/apps/snpeff/4.1g/snpEff.jar databases | grep -i <organism>
For example, if you are working with Arabidopsis thaliana:
java -jar /share/apps/snpeff/4.1g/snpEff.jar databases | grep -i thaliana
Output:
TAIR10.26 Arabidopsis_thaliana ENSEMBL_BFMPP_26_16 http://downloads.sourceforge.net/project/snpeff/databases/v4_1/snpEff_v4_1_ENSEMBL_BFMPP_26_16.zip
athaliana130 Arabidopsis_Thaliana http://downloads.sourceforge.net/project/snpeff/databases/v4_1/snpEff_v4_1_athaliana130.zip
athalianaTair10 Arabidopsis_Thaliana http://downloads.sourceforge.net/project/snpeff/databases/v4_1/snpEff_v4_1_athalianaTair10.zip
athalianaTair9 Arabidopsis_Thaliana http://downloads.sourceforge.net/project/snpeff/databases/v4_1/snpEff_v4_1_athalianaTair9.zip
Select the reference/version you are working with (for example: athalianaTair10), then:
java -jar snpEff.jar download athalianaTair10
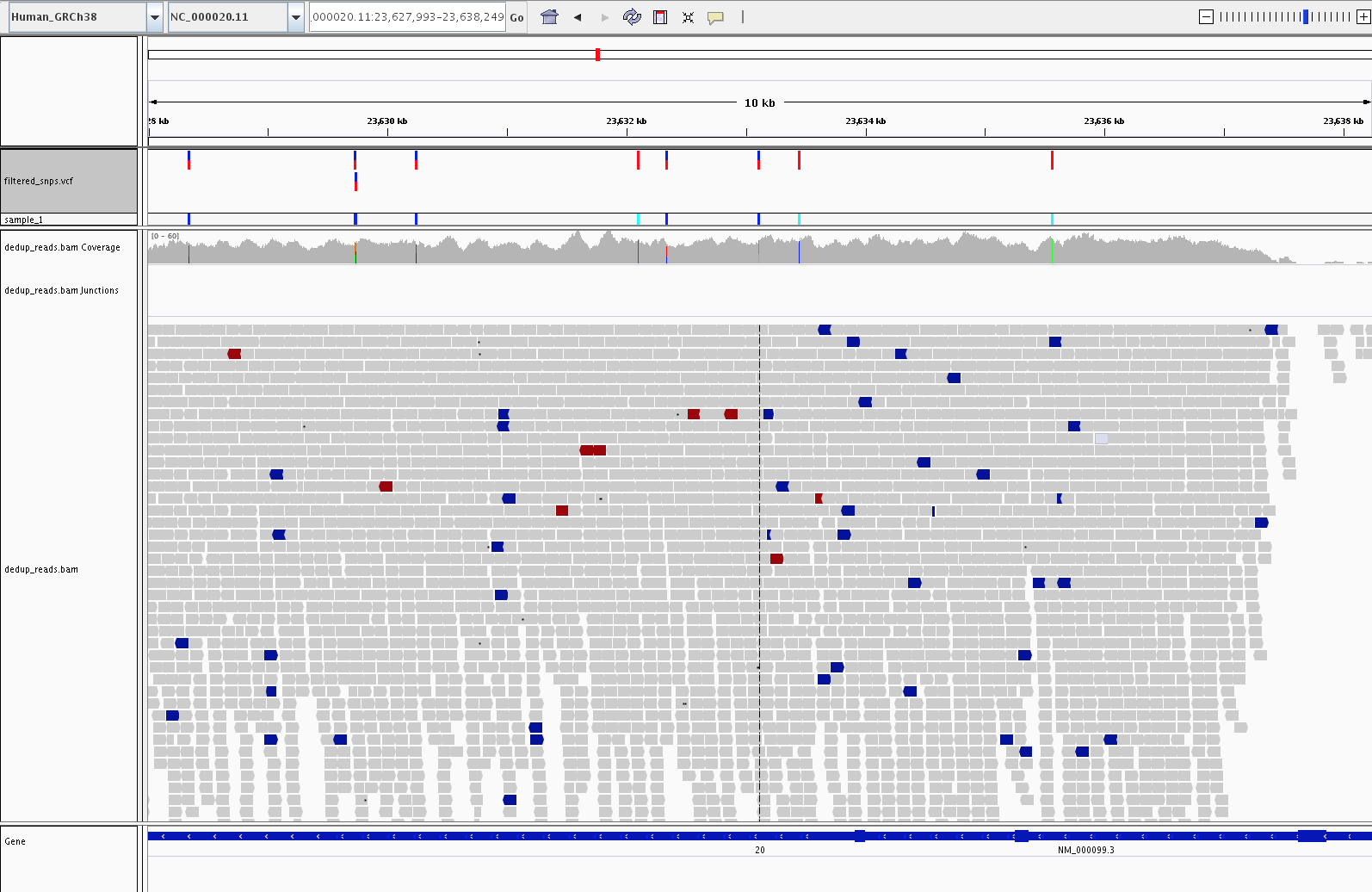
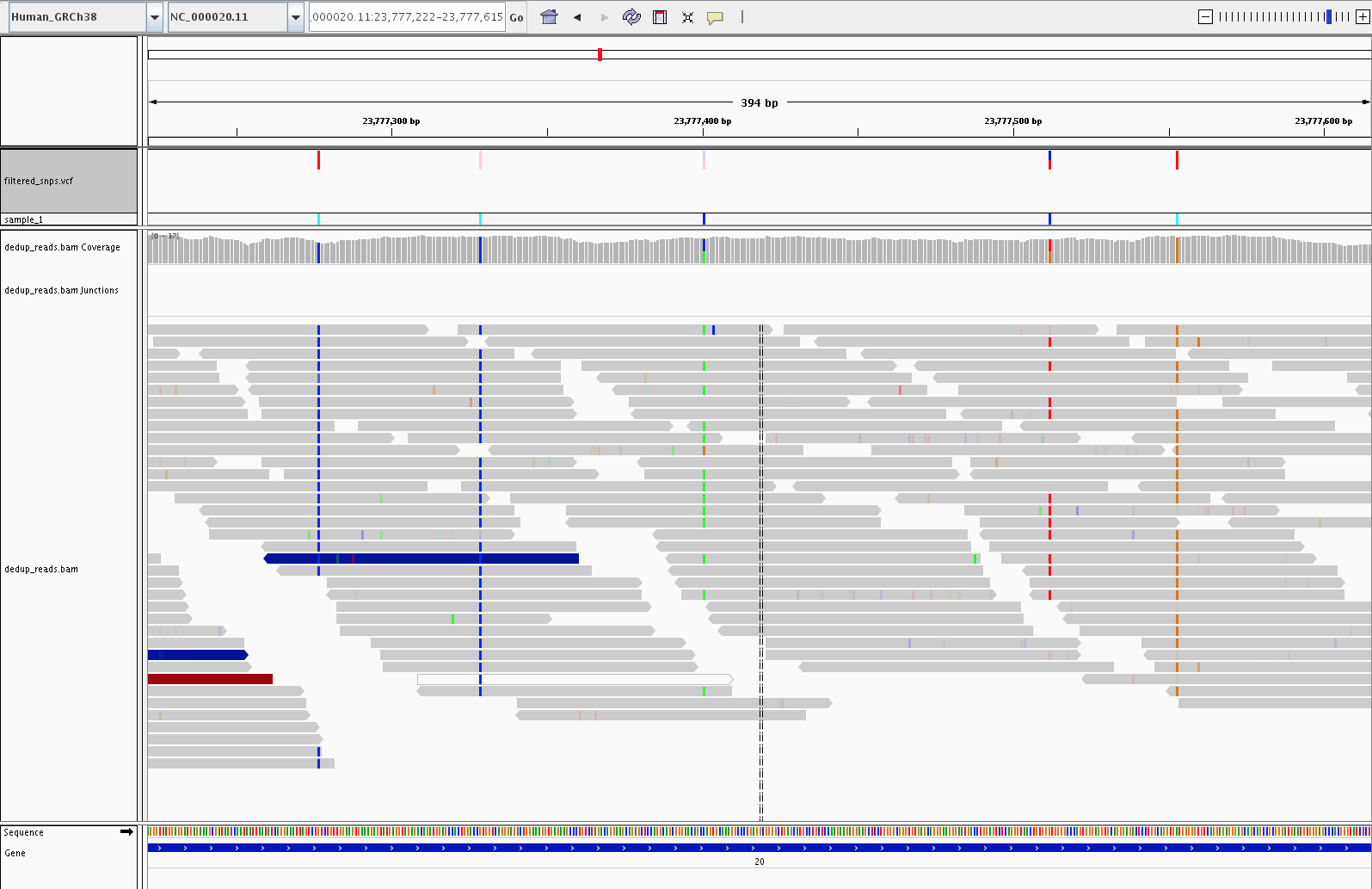
4) Visualization (IGV)
Let's fire up IGV, load the Human_GRCh38 genome, and load our dedup.bam and filtered_snps.vcf files. Let's look at:
1) Examples of high confidence SNPs in our filtered_snps.vcf file vs SNPs in our alignment not called by GATK

2) Examples of Homozygous variants vs Heterozygous variants.

3) Examples of SNPs filtered out by variant filtering (23, 703, 878)
4) Examples of SNPs in the cystatin C gene (CST3, variants in this gene are associated with Alzheimer's)